

磁珠提取结合液相色谱-串联质谱法检测尿游离儿茶酚胺及其代谢物的性能评价
目的
建立一种基于磁珠提取联合液相色谱-串联质谱法(LC-MS/MS)检测人尿液中儿茶酚胺及其代谢物的方法,并评价其方法学性能。
方法
基于磁珠提取前处理联合LC-MS/MS技术,采用ChromCore PFP色谱柱(2.1 mm×100 mm,3.0 μm),在柱温40 ℃ 条件下,以0.1%甲酸水-甲醇体系为流动相,流速0.3 ml/min,进行梯度洗脱。分离并测定人尿液中游离的肾上腺素(E)、去甲肾上腺素(NE)、多巴胺(DA)、甲氧基肾上腺素(MN)、甲氧基去甲肾上腺素(NMN)和3-甲氧基酪胺(3-MT)。同时系统评估该方法的线性、定量限、精密度、正确度、携带污染、基质效应及稳定性。收集2024年3月至2025年4月在遵义医科大学及各附属医院进行尿液儿茶酚胺检测的受试者及志愿者临床24 h尿液样本,共452例,用于建立参考区间。同时从中选取80份样本,分别采用磁珠提取和固相萃取法进行前处理,并通过LC-MS/MS检测。运用Passing-Bablok回归和Bland-Altman分析比较2种前处理方法结果的一致性。
结果
该方法通过了线性、定量限、精密度、正确度、携带污染、基质效应及稳定性性能评价。磁珠提取和固相萃取2种方法提取E、NE、DA、MN、NMN及3-MT斯皮尔曼等级相关系数均>0.99,存在强相关性,平均百分偏差均低于3%。参考区间分别为:E(0.71~16.97 μg/24 h)、NE(8.27~95.44 μg/24 h)、DA(54.00~408.66 μg/24 h)、MN(3.42~34.44 μg/24 h)、NMN(6.05~76.30 μg/24 h)、3-MT(10.08~65.66 μg/24 h)。
结论
磁珠提取尿液中儿茶酚胺及其代谢物操作简便,分析性能良好。
儿茶酚胺是一类由肾上腺髓质、交感神经元及肾上腺外嗜铬体分泌的含儿茶酚和胺基的神经类物质,主要包括肾上腺素(epinephrine,E)、去甲肾上腺素(norepinephrine,NE)和多巴胺(dopamine,DA),其在体内被儿茶酚-O-甲基转移酶分别代谢为甲氧基肾上腺素(metanephrine,MN)、甲氧基去甲肾上腺素(normetanephrine,NMN)和3-甲氧基酪胺(3-methoxytyramine,3-MT) [ 1 ] 。嗜铬细胞瘤/副神经节瘤(pheochromocytoma and paraganglioma,PPGL)是起源于肾上腺髓质或肾上腺外交感神经和副交感神经副神经节的罕见神经内分泌肿瘤,因过量分泌儿茶酚胺而导致继发性高血压等典型临床表现 [ 2 ] 。嗜铬细胞瘤和副神经节瘤诊断治疗专家共识(2020版)推荐,在PPGL的生化诊断中优先采用液相色谱-串联质谱法(liquid chromatography tandem mass spectrometry,LC-MS/MS)检测血浆游离或24 h尿液中的MNs(包括MN和NMN),同时建议联合检测E、NE、DA及其代谢物3-MT以提高诊断准确性 [ 3 ] 。在此背景下,随着LC-MS/MS技术日趋成熟及国产配套试剂盒获批数量持续增加,该技术凭借高灵敏度、高选择性和多指标同步检测的优势,已在临床儿茶酚胺检测领域获得广泛应用 [ 4 , 5 ] 。
当前尿液儿茶酚胺的样本前处理仍以固相萃取(solid phase extraction,SPE)为常用技术 [ 5 , 6 ] ,但该技术存在局限性,尤其是氮吹浓缩步骤操作耗时,且易导致样本回收率波动及操作者间变异系数升高。而基于自动化程度高、无需氮吹等技术优势,结合国产全自动提取设备的快速发展,磁珠提取(magnetic bead extraction,MBE)在儿茶酚胺提取领域得到逐步推广 [ 7 , 8 ] 。禹松林等 [ 8 ] 成功将MBE与LC-MS/MS联用,建立了血浆中MN、NMN和3-MT 3种代谢物的检测方法,结果显示该方法具有良好的检测性能。Han等 [ 9 ] 采用MBE结合高效液相色谱电化学检测技术,实现了E、NE和DA的有效测定,并获得了理想的回收率。然而,目前国内临床实验室采用MBE技术进行尿液样本自动化前处理应用较少,且鲜有文献对MBE联合LC-MS/MS技术同时测定24 h尿液中E、NE、DA、MN、NMN及3-MT这6种化合物的分析方法,开展系统性的方法学评价。基于上述现状,本研究拟采用MBE与LC-MS/MS联用技术,建立一种高灵敏度、自动化程度高的分析方法,实现尿液中E、NE、DA、MN、NMN及3-MT的同步准确定量,以期为临床尿儿茶酚胺检测提供新的技术参考。
一、对象
本研究纳入2024年3月至2025年4月在遵义医科大学及各附属医院接受尿液儿茶酚胺检测的受试者及志愿者452名(男性237名,女性215名),年龄18~85岁[53(41,63)岁]。纳入标准:年龄≥18岁,无明显机体功能障碍者。排除标准:尿液收集时间不符合24 h、未加酸酸化防腐尿液、未准确记录24 h总尿量者;患有肝肾疾病,长期服用抗抑郁药物者。本研究已通过遵义医科大学附属医院医学伦理委员会审批(KLLY-2024-052)。
二、方法
1.主要仪器与试剂耗材:高效液相色谱-串联质谱系统(品生医疗公司Qlife Lab 9000 Plus);Auto M32全自动样品处理系统(苏州艾捷博雅科技公司)。质谱级有机溶剂(甲醇、乙腈、异丙醇)和甲酸购自美国Fisher公司,冰乙酸购自阿拉丁公司,实验用水为娃哈哈纯净水。DA、3-MT及NE-d 6、MN-d 3内标购自美国Cerilliant公司,E购自德国Dr.Ehrenstorfer公司,NE、MN、NMN购自北京曼哈格生物科技公司,E-d 3、DA-d 4、NMN-d 3购自加拿大Toronto Research Chemicals公司,3-MT-d 4购自美国Cambridge Isotope Laboratories公司。实验耗材:ChromCore PFP色谱柱(2.1 mm×100 mm,3.0 μm)(苏州纳谱分析技术公司),PWCX 96孔固相萃取板和弱阳离子交换磁珠(苏州艾捷博雅科技有限公司)。
2.标本采集与预处理:所有研究对象在尿液采集前24 h内禁止服用任何药物、避免剧烈运动、禁酒,同时避免食用巧克力、香蕉、坚果等可能影响儿茶酚胺检测结果的食物,并保持正常的生活作息规律 [ 3 , 10 ] 。24 h尿液采集方案:受试者首次晨尿弃去并记录时间作为起点,随后24 h内所有尿液收集至含25 ml 50%乙酸的避光容器中,24 h后混匀尿液并记录总尿量,分装10 ml于尿管,-80 ℃保存待检。
3.标准品及内标品制备:精确称量各化合物标准品及内标1 mg,溶解于1 ml 0.1 mol/L盐酸-50%甲醇溶液中配制成1 mg/ml高浓度储备液,于-80 ℃冻存备用。采用0.1%甲酸水-0.1%抗坏血酸-乙腈溶液稀释储备液制备混合标准品储备液(E 1 μg/ml、NE 4 μg/ml、DA 20 μg/ml、MN 2 μg/ml、NMN 2 μg/ml、3-MT 4 μg/ml)和混合内标储备液(E-d 3 4 μg/ml、NE-d 6 20 μg/ml、DA-d 4 20 μg/ml、MN-d 3 2 μg/ml、NMN-d 3 2 μg/ml、3-MT-d 4 4 μg/ml)。使用0.01%抗坏血酸溶液通过梯度稀释法将混合标准品储备液稀释20、40、100、200、500、1 000、2 000、4 000倍制备系列标准溶液,将混合内标储备液稀释100倍制备内标工作液,所有溶液分装后于-80 ℃保存备用。
4.质控品制备:选取6名健康人24 h尿液经充分混匀后制备成混合尿样,并测定其本底值。通过加入不同浓度的标准品分别制备成低、中、高浓度质控品,分装后-80 ℃冰箱冻存备用。
5.MBE样本前处理:将弱阳离子交换磁珠(儿茶酚胺代谢产物的磁固相萃取快速检测方案——代谢组学磁珠-生物磁珠专家 - Purimag Bead 官网 | 高品质生物磁珠解决方案)用50%异丙醇溶液配制成50 mg/ml悬液。取50 μl待测样本,加入20 μl内标工作液和330 μl稀释液(15 mmol/L碳酸氢铵溶液),振荡混匀10 s后全部转移至96方孔工字板3(9)孔位,在1(7)孔位加入40 μl磁珠悬液( 表1 )。将前处理板置于M32全自动样本处理系统(苏州艾捷博雅科技公司)并运行程序,程序结束后收集6(12)孔位洗脱液,4 ℃ 21 100× g离心5 min,取50 μl上清液上机检测。

6.SPE样本前处理:PWCX 96 WellPlate SPE板依次用200 μl甲醇和200 μl超纯水活化后正压压干。取50 μl样本加入20 μl内标工作液和330 μl 15 mmol/L碳酸氢铵溶液,涡旋混匀10 s后全部上样,正压压干后依次用200 μl乙腈和200 μl超纯水淋洗并压干。采用80 μl含0.2%甲酸的80%乙腈洗脱液洗脱两次,合并洗脱液氮吹浓缩后,用80 μl 0.2%甲酸水复溶,振荡2 min,4 ℃ 21 100× g离心5 min,取50 μl上清液上机检测。
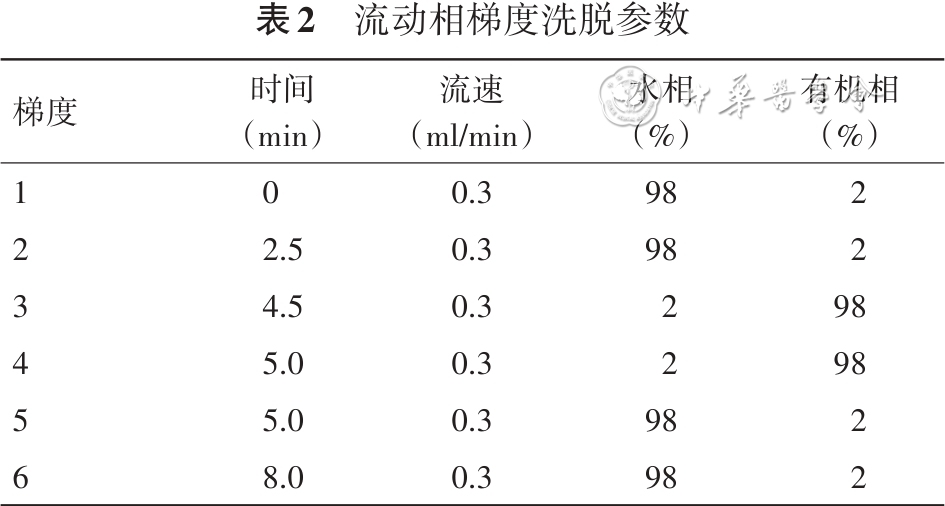
7.色谱分析条件:采用ChromCore PFP色谱柱(2.1×100 mm,3.0 μm),在柱温40 ℃ 条件下,以0.1%甲酸水-甲醇体系为流动相,流速 0.3 ml/min,进行梯度洗脱。流动相梯度详见 表2 。
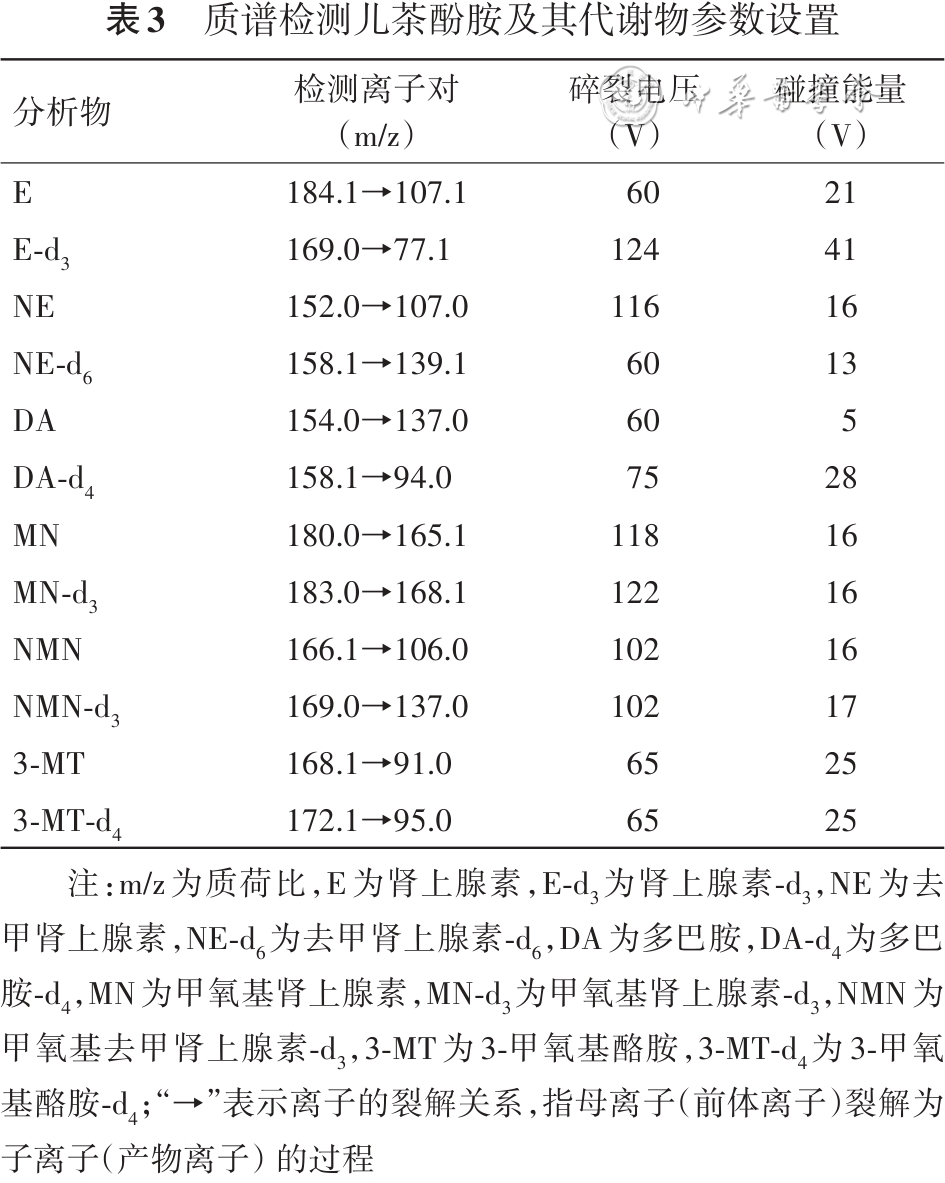
8.质谱分析条件:采用正离子电喷雾(ESI +)离子化方式结合多反应监测(MRM)模式,进样量1 μl,主要参数见 表3 。
三、方法学评价与验证
方法学评价参考《液相色谱-质谱临床应用建议》 [ 11 ] 《液相色谱串联质谱临床检测方法的开发与验证》 [ 12 ] 及CLSI C62-A [ 13 ] 建议。
1.线性评估:采用梯度稀释法将混合标准品储备液分别稀释20、40、100、200、500、1 000、2 000和4 000倍,制备8个浓度梯度的标准曲线工作液。每个浓度点进行3次重复检测,要求各浓度点的检测偏倚均在±15%范围内。标准曲线的建立以各浓度点标准品浓度为横坐标,目标物峰面积与对应内标峰面积的比值为纵坐标,采用加权最小二乘法(权重系数1/ x²)进行线性回归分析,相关系数( r)>0.99判定为标准曲线线性良好。
2.定量限:将低浓度标准溶液稀释至接近方法线性定量下限的浓度水平,制备为测试样品。每个浓度样品分装15份,按每批次5份的规格分3批完成检测。以信噪比(S/N)>10、总精密度≤20%且测定均值与理论值偏差≤15%的最低浓度作为方法的定量限。
3.精密度评价:检测低、中、高浓度质控品,每个浓度检测3个批次,每批次5个平行样本,分别计算不同浓度质控品批内和连续3 d批间的变异系数(coefficient of variation, CV),要求批内和批间 CV<15%。
4.正确度:通过加标回收实验评估方法的正确度,低、中、高质控样本各5份,每份各测3次。计算公式:加标回收率=(测量浓度-本底浓度)/加标浓度×100%,回收率在85%~115%判断为可接受。
5.携带污染:首先重复进样标准曲线的低浓度样本(定量下限),然后交替进样标准曲线的高浓度样本(定量上限)和低浓度样本(高低模式重复5个循环)。携带污染率=(高浓度后低浓度样本均值-连续低浓度样本均值)/连续低浓度样本均值×100%。若携带污染率<20%判断为可接受。
6.基质效应:选取6名健康人24 h尿液混合后均质分装20份,经MBE前处理后合并上清液作为空白基质。分别添加标准品配制低、中、高浓度质控品,同时制备相应浓度的纯溶剂对照。各浓度点设置3个平行样本,每样本重复测定3次(共9个数据点)。计算公式:基质效应(%)=目标物[(质控品响应值-空白基质响应值)/纯溶剂响应值]/(质控品内标响应值/纯溶剂内标响应值)×100%。要求各浓度点的基质效应值均应在85%~115%。
7.稳定性实验:评估低、高浓度质控品在生物基质中的稳定性,包括样本处理前和处理后进样盘放置的稳定性。将处理前的质控品分别放在室温(24和72 h)、4 ℃(24和72 h)条件下保存后检测;冻融稳定性测试采用-80 ℃保存经过3次冻融循环后检测(每次冻融循环间隔24 h);样本处理前稳定性计算样本放置后与放置前测定的浓度偏差;处理后样本计算4 ℃放置24、72 h与首次检测浓度的偏差。稳定性实验均设置3个平行样本。
四、建立参考区间
采用Dixon法识别并剔除离群值。无论数据是否服从正态分布,均采用非参数法进行统计分析。表观健康人群根据性别分组,并运用非参数秩和检验分析是否存在差异,存在差异则分组建立参考区间。采用Spearman相关性分析判断年龄是否与分析物存在相关性,存在相关性则按年龄分段建立参考区间。采用第2.5百分位数和第97.5百分位数作为参考区间的上下限,以( P 2.5, P 97.5)表示。
五、方法学比对
依据CLSI EP09C指南,本研究对80份临床24 h尿液儿茶酚胺样本开展方法学比对。样本经实验室自建的SPE方法和本研究的MBE方法分别前处理后,使用同一LC-MS/MS平台检测,并采用Bland-Altman分析和Passing-Bablok回归评估2种方法间的差异和相关性,Spearman等级相关系数(spearman rank correlation coefficient, r)越接近1,表明相关性越强 [ 14 , 15 ] 。
六、统计学分析
使用SPSS 29.0软件进行统计学处理及数据分析,计算均值、标准差、 CV、偏差和回收率。使用Origin 2024软件绘制色谱图,使用Medcalc 22.0绘制Passing-Bablok回归图和Bland-Altman差异图,进行方法学比对。 P<0.05为差异有统计学意义。
一、色谱图
尿液样本经MBE提取后,6种目标分析物(E、NE、DA、MN、NMN及3-MT)峰形良好,保留时间(min)分别为:1.99、1.29、2.84、5.37、2.47、5.94,分析物典型色谱图见 图1 。
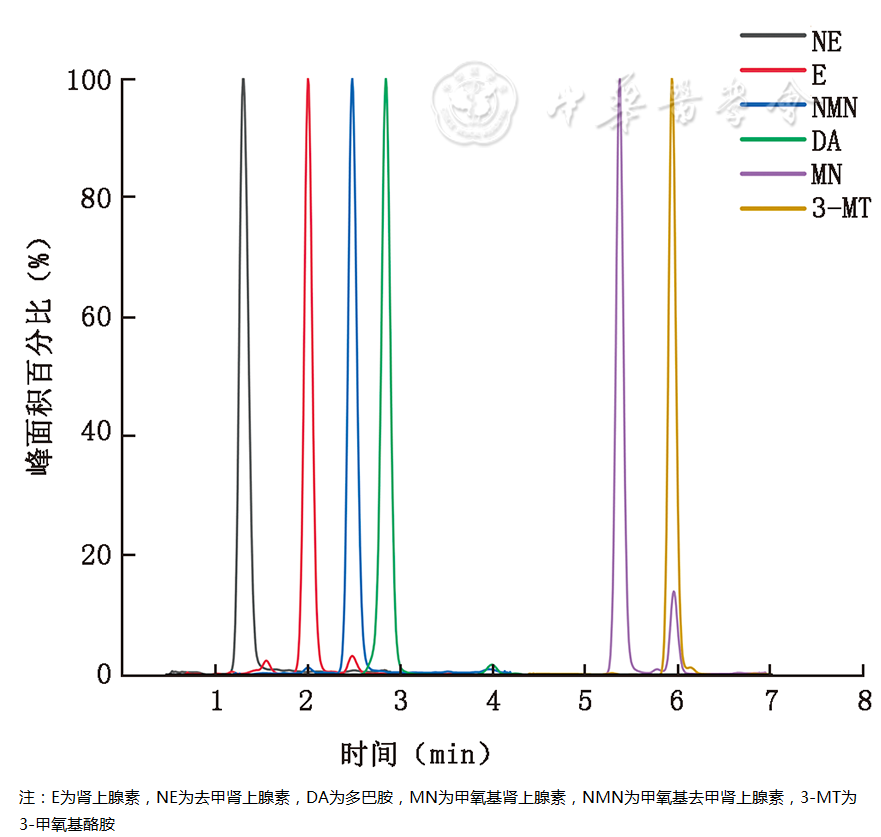
图1 尿液样本经磁珠提取后的典型色谱峰图
二、方法学评价
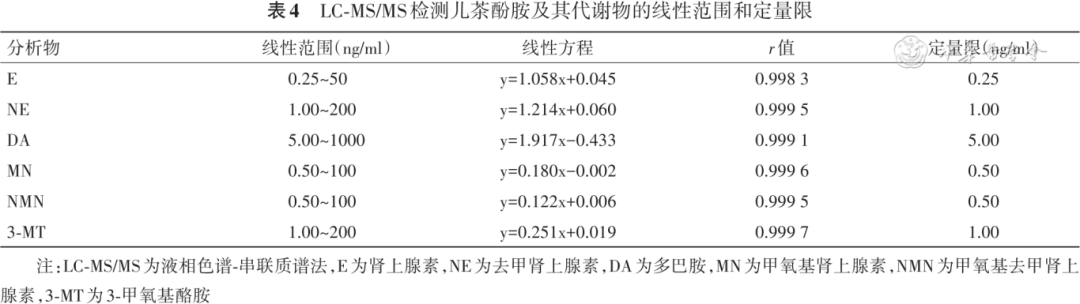
1.线性评估与定量限:在6种分析物的8个浓度点中,拟合标准曲线的 r值均>0.990,且定量限满足临床检测需求( 表4 )。
2.精密度:6种分析物的批内 CV范围为0.83%~4.80%,批间 CV范围为0.82%~4.05%,所有测定结果均满足方法学验证中 CV<15%的标准要求( 表5 )。
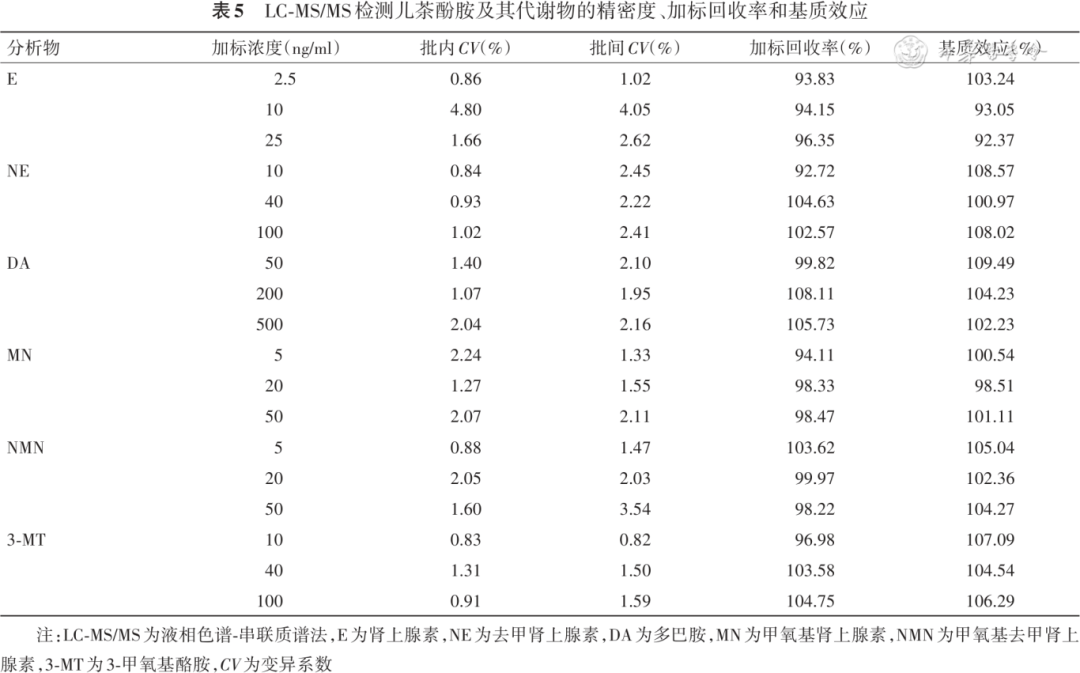
3.正确度:通过加标回收率实验评估方法的正确度。结果显示,6种分析物的回收率范围为92.72%~108.11%,均满足回收率在100%±15%的要求( 表5 )。
4.基质效应:经同位素内标校正后,6种分析物的相对基质效应均处于100%±15%的可接受范围内,E(92.37%~103.24%)、NE(100.97%~108.57%)、DA(102.23%~109.49%)、MN(98.51%~101.11%)、NMN(102.36%~105.04%)和3-MT(104.54%~107.09%)( 表5 )。
5.携带污染:E、NE、DA、MN、NMN及3-MT的携带污染率分别为-3.84%、8.02%、5.01%、-2.31%、2.32%、3.97%,6种分析物的携带污染率均低于20%的允许标准。
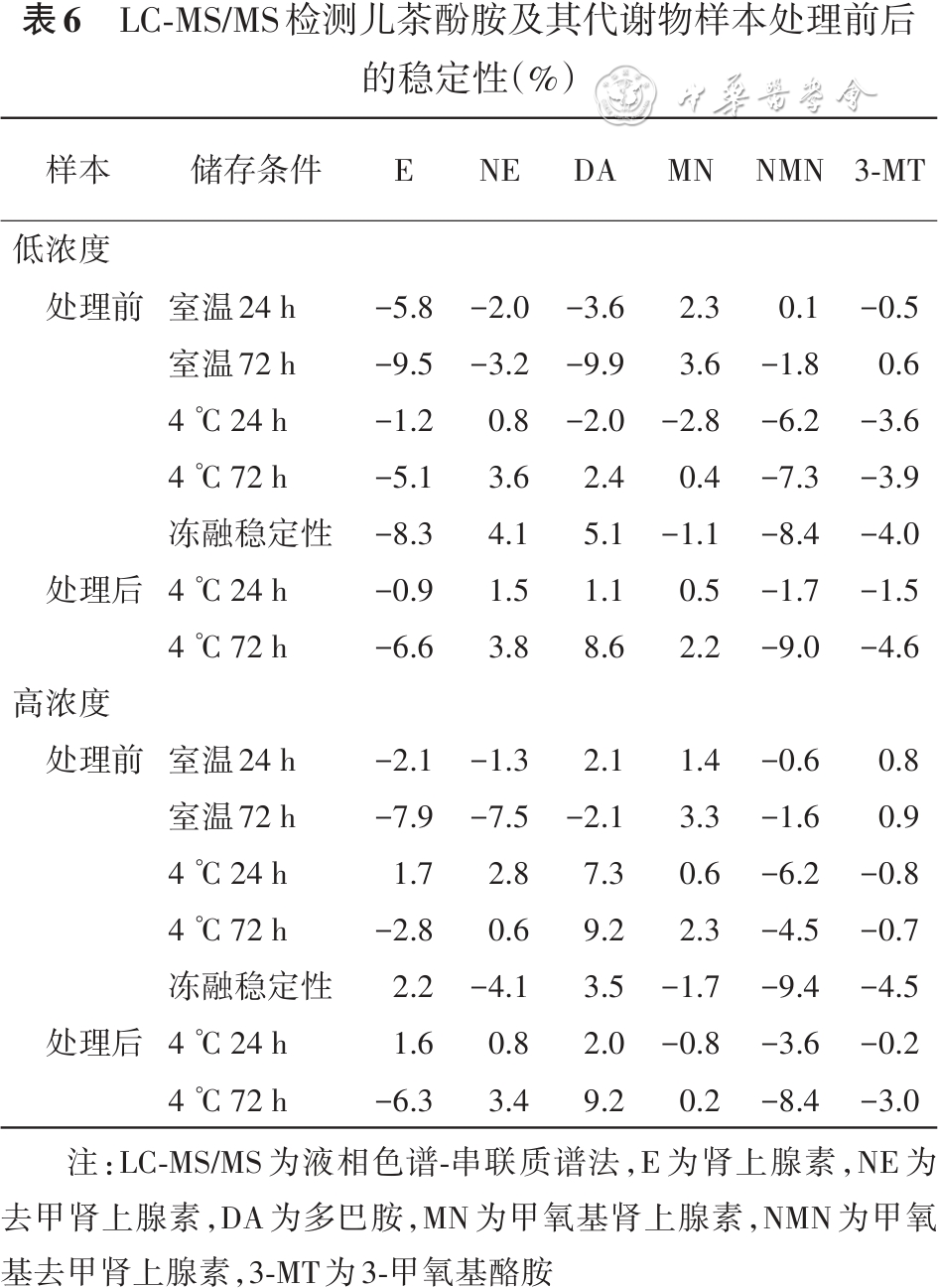
6.稳定性:6种分析物的稳定性评估结果显示,样本处理前的稳定性偏差分别为:E(-9.5%~2.2%)、NE(-7.5%~4.1%)、DA(-9.9%~9.2%)、MN(-2.8%~3.6%)、NMN(-9.4%~0.1%)和3-MT(-4.5%~0.9%);样本处理后的稳定性偏差分别为:E(-6.6%~1.6%)、NE(0.8%~3.8%)、DA(1.1%~9.2%)、MN(-0.8%~2.2%)、NMN(-9.0%~-1.7%)和3-MT(-4.6%~-0.2%)。所有测试条件下的结果偏差均处于±15%的可接受标准内( 表6 )。
三、参考区间
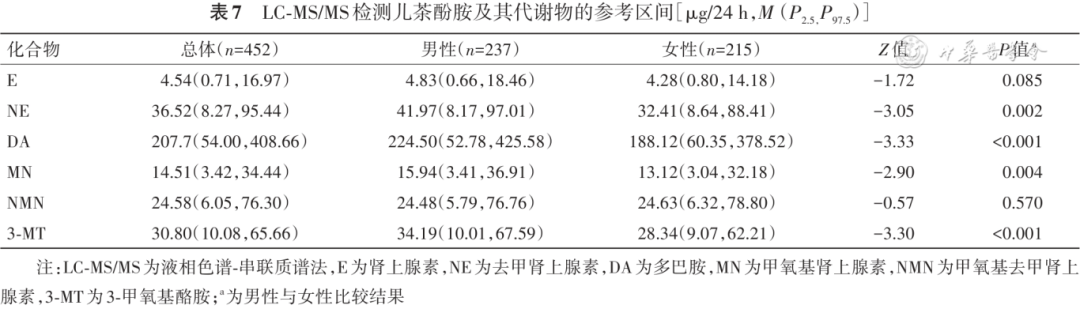
共纳入452名表观健康者,尿液中E、NE、DA、MN、NMN及3-MT水平均与年龄无相关性( r s 为-0.034~0.085, P均>0.05)。此外,男性与女性在E及NMN水平上差异也无统计学意义( P均>0.05)( 表7 )。
四、方法学比对
通过检测值乘以24 h总尿量将浓度单位(ng/ml)转换为总量单位(μg/24 h)进行方法学比较。Bland-Altman分析结果显示( 图2 ),MBE与SPE测定结果的平均偏差分别为:E(0.9%)、NE(2.0%)、DA(0.3%)、MN(2.0%)、NMN(-0.7%)及3-MT(-1.3%),6种分析物的偏差值落在±1.96SD外的比例均<5%。Spearman相关性分析表明,2种方法测定结果均有良好的相关性( r均>0.99, P均<0.001),E、NE、DA、MN、NMN及3-MT 的 r值分别为0.998、0.994、0.999、0.993、0.997、0.993。
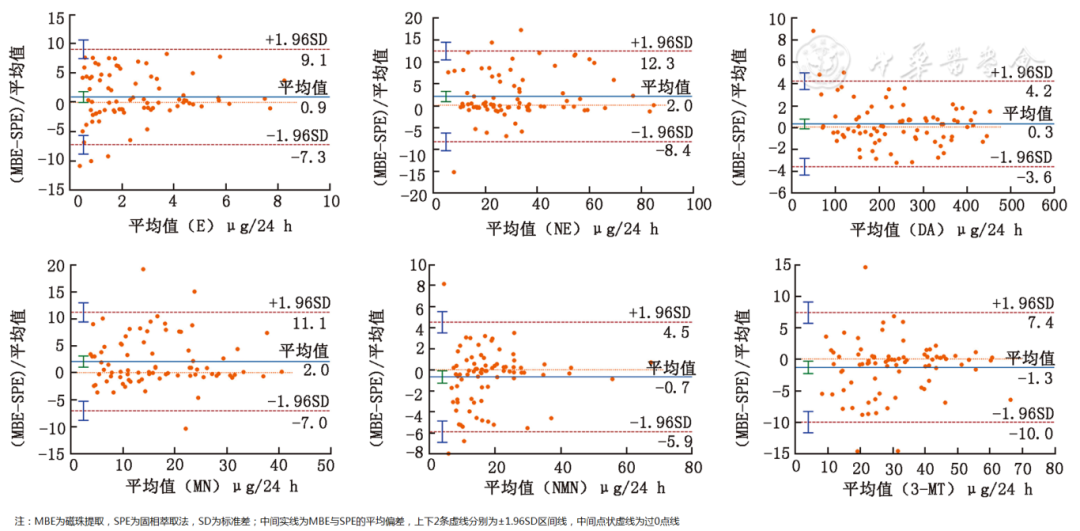
图2 80份临床样本MBE和SPE提取儿茶酚胺及其代谢物检测结果一致性比对Bland-Altman图
讨 论
儿茶酚胺及其代谢物的实验室检测已明确用于PPGL的辅助诊断,其中血浆样本因采集便利性及良好的特异性和敏感性成为常用检测类型,但其要求卧位休息30 min后采血以降低假阳性,这不便于门诊患者 [ 16 ] 。而24 h尿液检测不受采集体位影响,且分析物浓度较高,更易于建立稳定的检测方法。尿液中儿茶酚胺及其代谢物以2种形式存在——游离型和经磺基转移酶1A3催化4-羟基硫酸化生成的硫酸结合型,两者合称为分馏型 [ 17 , 18 ] 。其中尿液MNs和3-MT以结合型为主,而E、NE、DA以游离型为主 [ 19 ] 。虽然美国内分泌学会2014年发布的PPGL诊断指南推荐检测24 h尿分馏型MN [ 20 ] ,但在国内临床实践中,采用分馏型MNs检测尚未达成共识,尿游离型MN检测反而更为普遍。王恺隽等 [ 17 ] 通过加浓盐酸酸化并沸水浴30 min将分馏型MN转化为游离形式,与未加酸水解的游离型分别进行检测评估对嗜铬细胞瘤的诊断效能,发现尿游离型MN对嗜铬细胞瘤的诊断效能与分馏型基本相当。Eisenhofer等 [ 21 ] 指出与尿分馏型相比,尿游离型MN的测定具有更高的诊断特异性和阳性预测值,且对于嗜铬细胞瘤低危人群与血浆MN相比诊断优势相当。此外,与分馏型相比,游离型儿茶酚胺及其代谢物的检测无需浓盐酸水解及沸水浴步骤,操作流程更为简便且检测结果受富含儿茶酚胺的饮食影响更小 [ 22 ] 。然而目前国内基于尿游离儿茶酚胺及其代谢物的参考区间研究较少,且梅奥诊所实验室提供的MN参考区间不符合游离型范畴。我们通过纳入≥18岁表观健康人群所建立的参考区间显示,24 h尿游离E、NE、DA、MN、NMN及3-MT的水平与年龄均无相关性,这不同于血浆游离NMN和3-MT随年龄增长而增高 [ 23 ] ,其可能是因为血浆NMN的年龄依赖性升高主要由交感神经活性增强驱动,而3-MT的升高与DA代谢增强及清除率下降相关 [ 24 ] 。本研究的参考区间是基于贵州地区人群建立,其饮食习惯、环境因素等方面的差异可能影响研究结果的普适性。因此,有待进一步开展多中心、跨地域的大样本研究与验证。
高效液相色谱因其高分离效率,被广泛用于儿茶酚胺及其代谢物分析,常与电化学 [ 9 ] 、荧光 [ 25 , 26 ] 或质谱检测器联用实现定量检测。Hu等 [ 25 , 26 ] 通过MBE结合高效液相色谱-荧光检测法,开发了灵敏的尿液E、NE及DA分析方法,虽具有良好的线性和灵敏度,但在分子覆盖范围和特异性上仍有限。而LC-MS/MS 具备更广的分子覆盖范围和更高的特异性,然而其分析性能在很大程度上依赖于样品前处理的优化,其优化程度直接影响目标分析物的回收率和检测可靠性。目前常用的前处理方法主要包括SPE [ 5 , 6 ] 、MBE [ 8 , 27 ] 、蛋白沉淀法 [ 28 ] 、液液萃取法 [ 29 ] 、稀释法 [ 4 ] 等。在尿液儿茶酚胺检测中,虽然样本浓度较高可采用最简单的稀释法,但该方法存在局限性:一方面,稀释处理会增强基质效应并引入杂峰干扰,常导致关键分析物如E和NE未检出;另一方面,长期采用会加速色谱柱性能衰减,表现为柱效下降和保留时间漂移等问题 [ 4 ] 。相较于MBE,SPE虽能提高检测灵敏度,但普遍存在操作流程繁琐、耗材成本高(本研究中单个样本的提取耗材成本超过MBE方法的3倍)且对实验人员技术要求较高。
本研究基于LC-MS/MS技术平台结合MBE建立了一种可同时定量尿液中E、NE、DA、MN、NMN及3-MT的分析方法。方法采用的超顺磁性磁珠,其高磁响应特性使样品分离富集过程缩短至约8 min,结合自动化设备,实现了磁珠的活化、平衡、淋洗、洗脱等环节的自动一体化操作,减少了人工干预和操作误差,有效提升了方法的可重复性和批量检测能力。我们通过优化流动相梯度后色谱分析结果显示,6种分析物在6 min内均实现良好分离,同时方法学评价显示我们建立的方法性能良好。但本方法仍存在一定不足:(1)前处理流程中试剂添加和样本转移步骤仍需人工干预;(2)单次样本提取通量仍较低(32份/批);(3)洗脱阶段存在磁珠共洗脱风险,需增加离心步骤以防色谱柱堵塞。后续研究将着力开发全自动加样模块并扩大处理通量。综上所述,本研究证实,磁珠提取技术为LC-MS/MS前处理提供了新型自动化解决方案,其通过高效富集E、NE、DA、MN、NMN及3-MT分析物,显著提升了尿液儿茶酚胺及其代谢物的提取效率,为实验室开展高通量检测提供了可靠的技术支持。
- 上一篇:核酸提取解析:方法、工作流程及最佳实践 2026/6/17
- 下一篇:磁免疫化学发光试剂研发过程中固相载体磁性微球的选择指南 2026/3/25